Submitted by M.T. Ruggiero on Wed, 02/11/2016 - 13:52
The work showcases the ability of terahertz spectroscopy to probe temperature dependant changes in the low-frequency vibrational modes, and ultimately relates them to intermolecular anharmonicity.
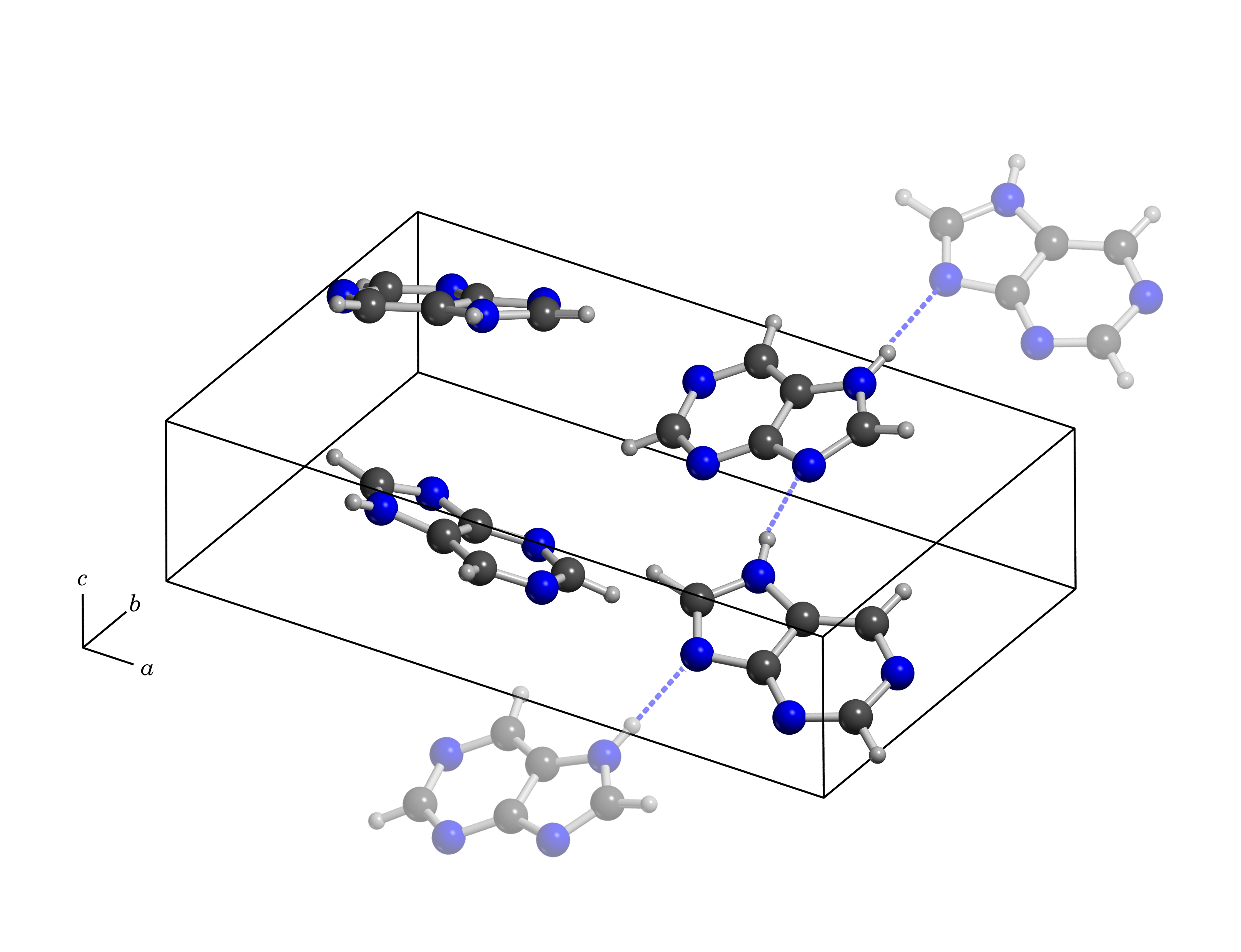
The research, published as Resolving the Origins of Crystalline Anharmonicity using Terahertz Time-Domain Spectroscopy and ab initio Simulations was recently published in the Journal of Physical Chemistry B. In it, members of the TAG group were able to show that the low-frequency vibrations, which are highly dependant on the subtle shape of the intermolecular potential energy surface, are able to help resolve deviations from strictly harmonic interactions.
The experimental data was coupled with two theoretical techniques, solid-state density functional theory (DFT) and ab initio molecular dynamics (AIMD). While DFT methods have been used extensively to describe the vibrational properties of materials, they are not able to take temperature into account and thus do not capture anharmonic effects, but they yield very detailed information about the vibrational motions. On the other hand, AIMD simulations can account for thermal differences, but they lack the precise vibrational data provided by DFT. Here, these two methods were combined in an attempt to understand the vibrational dynamics of crystalline purine, the building block of many DNA bases.
This exciting work can be viewed here.